Aperçu: G.M.
Résumé
Une activité stéroïdogène prénatale latente élevée a été observée dans le liquide amniotique de garçons autistes, sur la base de la mesure des androgènes prénatals et d’autres hormones stéroïdiennes. À ce jour, il n'est pas clairement établi si d'autres stéroïdes prénataux contribuent également à la probabilité d'autisme. Les œstrogènes prénataux doivent être étudiés car ils jouent un rôle clé dans la synaptogenèse et la corticogenèse au cours du développement prénatal, chez les hommes comme chez les femmes. Ici, nous testons si les niveaux prénataux d'oestriol, d'œstradiol, d'œstrone et de sulfate d'œstrone dans le liquide amniotique sont associés à l'autisme, dans la même cohorte de naissance historique danoise, dans laquelle les androgènes prénataux ont été mesurés à l'aide d'une régression logistique univariée (n = 98 cas, n = 177 contrôles). Nous faisons également une comparaison semblable entre les œstrogènes et les androgènes prénatals. L'œstradiol, l'œstrone, l'oestriol et la progestérone sont tous deux liés à l'autisme dans les analyses univariées après correction avec un taux de fausse découverte. Une comparaison des rapports de cotes standardisés a montré que l'œstradiol, l'œstrone et la progestérone avaient les effets les plus importants sur la probabilité d'autisme. Ces résultats montrent pour la première fois que les œstrogènes prénataux contribuent à la probabilité de l'autisme, en prolongeant la découverte d'une activité stéroïdogène prénatale élevée dans l'autisme. Cela affecte probablement la différenciation sexuelle, le développement et la fonction du cerveau.
Une activité stéroïdogène prénatale latente élevée a été observée dans le liquide amniotique de garçons autistes, sur la base de la mesure des androgènes prénatals et d’autres hormones stéroïdiennes. À ce jour, il n'est pas clairement établi si d'autres stéroïdes prénataux contribuent également à la probabilité d'autisme. Les œstrogènes prénataux doivent être étudiés car ils jouent un rôle clé dans la synaptogenèse et la corticogenèse au cours du développement prénatal, chez les hommes comme chez les femmes. Ici, nous testons si les niveaux prénataux d'oestriol, d'œstradiol, d'œstrone et de sulfate d'œstrone dans le liquide amniotique sont associés à l'autisme, dans la même cohorte de naissance historique danoise, dans laquelle les androgènes prénataux ont été mesurés à l'aide d'une régression logistique univariée (n = 98 cas, n = 177 contrôles). Nous faisons également une comparaison semblable entre les œstrogènes et les androgènes prénatals. L'œstradiol, l'œstrone, l'oestriol et la progestérone sont tous deux liés à l'autisme dans les analyses univariées après correction avec un taux de fausse découverte. Une comparaison des rapports de cotes standardisés a montré que l'œstradiol, l'œstrone et la progestérone avaient les effets les plus importants sur la probabilité d'autisme. Ces résultats montrent pour la première fois que les œstrogènes prénataux contribuent à la probabilité de l'autisme, en prolongeant la découverte d'une activité stéroïdogène prénatale élevée dans l'autisme. Cela affecte probablement la différenciation sexuelle, le développement et la fonction du cerveau.
Introduction
La prévalence de l'autisme axée sur les hommes [1, 2], ainsi que la découverte selon laquelle les filles autistes ont une charge mutationnelle supérieure à celle des garçons autistes [3,4,5], suggèrent que les hommes sont plus susceptibles de développer l'autisme. Le sex-ratio dans les diagnostics d'autisme persiste même après la prise en compte d'un sous-diagnostic et / ou d'un diagnostic erroné, ainsi que d'un camouflage chez les femmes, les hommes étant trois fois plus susceptibles de souffrir du trouble [6]. Cela implique des mécanismes de différenciation sexuelle dans le développement de l'autisme. Cinq conclusions récentes appuient cette inférence.
- Premièrement, les femmes autistes ont une structure cérébrale atypique dans les régions dimorphes sexuellement, évaluée par imagerie par résonance magnétique et comparée à des contrôles neurotypiques [7].
- Deuxièmement, la connectivité fonctionnelle dans le cerveau des hommes autistes présente à la fois des profils hyper-masculin et hyper-féminin, lorsqu'elle est évaluée par rapport aux différences neurotypiques entre les sexes [8].
- Troisièmement, les personnes autistes montrent un changement de masculinisation des scores sur deux traits psychologiques dimorphes sexuels majeurs, l'empathie et la systématisation, constat qui a été reproduit dans une grande étude de données portant sur 36 000 personnes autistes [9, 10].
- Quatrièmement, les femmes autistes ont un taux élevé d'androstènedione, précurseur de la testostérone [11].
- Enfin, cinquièmement, l'enfant autiste présente des traits faciaux hyper-masculinaux évalués par photogrammétrie tridimensionnelle [12].
Bien que l’autisme soit fortement héréditaire et que des mécanismes génétiques liés au sexe puissent contribuer à cette implication de la différenciation sexuelle dans l’autisme [2, 5], l’exposition prénatale aux hormones et une brève augmentation de la testostérone fœtale sont essentielles à la différenciation sexuelle et à la masculinisation chez l’être humain [13, 14 ]. Dans le même esprit, nous avons précédemment constaté une activité stéroïdogène élevée au cours de cette fenêtre de masculinisation prénatale (PMW) dans le liquide amniotique de garçons autistes [15]. Par la suite, trois très grandes études épidémiologiques ont révélé un lien entre l'autisme et le syndrome des ovaires polykystiques (SOPK) de la mère, une affection associée à un excès androgène [16,17,18].
Conformément à cela, le ratio de chiffres 2D: 4D, un marqueur de l'exposition prénatale aux androgènes, est également masculinisé chez les enfants autistes et leurs parents [19]. Enfin, les femmes autistes et leurs mères présentent des taux élevés de cancers liés aux stéroïdes, tels que le cancer du sein et le cancer de l'ovaire [20].
Cependant, un certain nombre d'études axées sur la testostérone n'ont pas reproduit la corrélation entre les niveaux hormonaux et les traits autistiques.
Cependant, un certain nombre d'études axées sur la testostérone n'ont pas reproduit la corrélation entre les niveaux hormonaux et les traits autistiques.
Premièrement, la testostérone du cordon ombilical mesurée peu après la naissance n'était pas associée au développement de traits autistiques [21].
Deuxièmement, la testostérone salivaire au cours d’une brève période d’apparition de stéroïdes postnatales («mini-puberté») n’a pas non plus de corrélation avec les traits autistiques chez les tout-petits [22].
Dans les deux cas, la testostérone a été mesurée après la naissance - pendant la période néonatale - plutôt que pendant la PMW, au cours duquel la testostérone fœtale est produite pour la première fois et la masculinisation du cerveau et du corps commence. Cela suggérerait que le timing est crucial pour les effets de la testostérone sur le cerveau, la PMW tardive au début du début du deuxième trimestre étant la clé, plutôt que la période néonatale. Enfin, l'évaluation univariée de la testostérone amniotique dans une cohorte distincte d'enfants neurotypiques n'a pas non plus révélé d'association à des traits autistiques chez l'enfant [23].
Cette dernière conclusion peut refléter le fait que l'environnement endocrinien au sens large en dehors de la testostérone est également important pour la probabilité de l'autisme.
Discussion
Cette étude rapporte les premières preuves que des niveaux élevés d'estradiol, d'oestriol et d'œstrone amniotiques prénatals sont associés à l'autisme, les taux d'œstradiol étant le facteur prédictif le plus significatif de la probabilité d'autisme dans les modèles de régression logistique univarié.
Ces découvertes complètent les observations antérieures selon lesquelles une activité stéroïdogène élevée est associée à l'autisme dans les mêmes échantillons provenant de la cohorte de naissance historique danoise [15].
Nous avons également calculé des OR (Odd Ratio) normalisés, afin de comparer directement les tailles d'effet de tous les stéroïdes amniotiques mesurés à ce jour.
Nous avons constaté que l'œstradiol avait l'effet d'effet positif le plus important sur la probabilité d'autisme, suivi de l'œstrone, de l'oestriol et de la progestérone (Fig. 3).
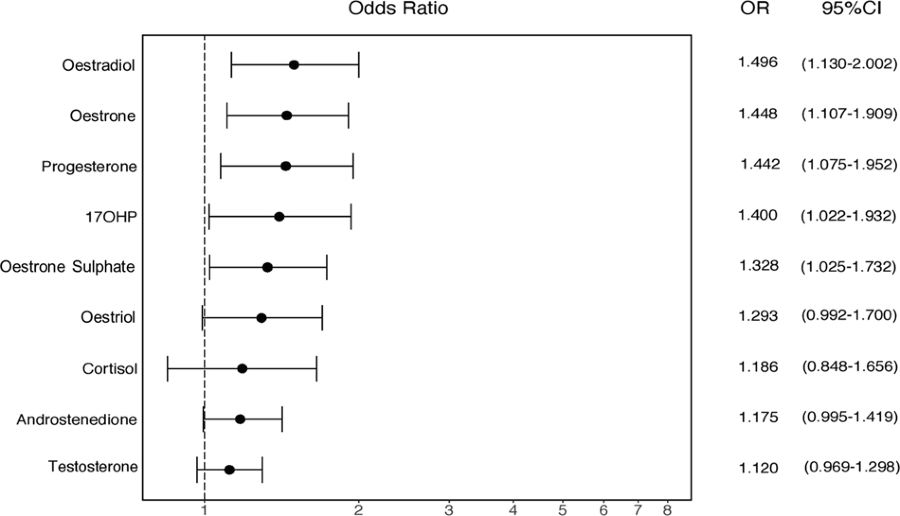
Cette constatation semble contredire un rapport précédent de Windham et al. [32] qui ont montré que les concentrations plus faibles d'oestriol au deuxième trimestre étaient modestement associées à un diagnostic ultérieur d'autisme chez la progéniture. Cependant, nos échantillons correspondent à un moment légèrement plus précoce de la grossesse par rapport à Windham et al. (semaine de gestation moyenne = 14,9 vs 17,2 respectivement) (voir tableau 1) [32], ce qui pourrait potentiellement mieux capturer la poussée de stéroïdes au cours de la PMW [14]. De plus, nos échantillons sont d’origine différente, puisque Windham et al. ont dosé le sérum maternel, plutôt que le liquide amniotique fœtal. Les taux d'hormones stéroïdes dans le sérum maternel ne diffèrent pas du sexe du bébé et ne sont pas en corrélation avec les niveaux amniotiques au cours du PMW [42]. Par conséquent, les œstrogènes amniotiques sont sans doute plus pertinents pour la question de recherche actuelle que les œstrogènes sériques maternels.
Une différence de taux d'œstrogènes entre la mère et l'enfant pourrait potentiellement être attribuée au placenta, qui agit comme régulateur endocrinien de l'interface mère-fœtus et principale source de production d'œstrogènes pour le fœtus via l'aromatisation d'androgènes [43]. Plusieurs sources de données suggèrent un rôle contributif du placenta dans l’étiologie de l’autisme.
Une différence de taux d'œstrogènes entre la mère et l'enfant pourrait potentiellement être attribuée au placenta, qui agit comme régulateur endocrinien de l'interface mère-fœtus et principale source de production d'œstrogènes pour le fœtus via l'aromatisation d'androgènes [43]. Plusieurs sources de données suggèrent un rôle contributif du placenta dans l’étiologie de l’autisme.
Premièrement, il y a une augmentation de l'inflammation placentaire dans l'autisme [44].
Deuxièmement, il existe une morphologie atypique du placenta [45] et une augmentation de la taille du placenta [46] en cas d'autisme et à risque familial élevé, respectivement.
Troisièmement, les complications liées au placenta (pré-éclampsie [47], troubles hypertensifs [48]) sont également plus fréquentes lors des grossesses menant à l'autisme.
Comme pour l'autisme, le dysfonctionnement placentaire affecte également de manière disproportionnée les hommes plus que les femmes [49].
Étant donné les corrélations par paires élevées entre de nombreuses hormones stéroïdiennes (Fig. 2, tableau supplémentaire 3),
Étant donné les corrélations par paires élevées entre de nombreuses hormones stéroïdiennes (Fig. 2, tableau supplémentaire 3),
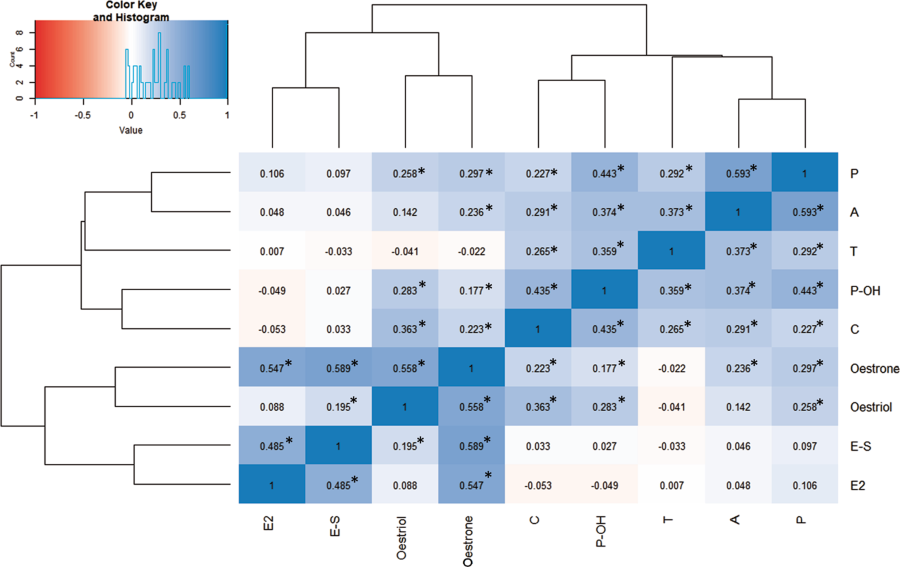
ainsi que l'absence de différence d'aromatisation entre les cas et les témoins, nos données suggèrent qu'une augmentation des œstrogènes fœtaux est secondaire à une activité accrue de l'intégralité de l'axe stéroïdogène dans les grossesses aboutissant plus tard à l'autisme [15].
Il est intéressant de noter que l’œstradiol n’était pas significativement corrélé à la testostérone (β = 0,007 de Pearson, p = 0,9103) malgré leur proximité dans la stéroïdogenèse. Cette différence peut être due au fait que les œstrogènes sont également produits de novo par le placenta, en plus d'être aromatisés à partir d'androgènes fœtaux et maternels [43, 50]. Ainsi, une approche multi-systèmes est nécessaire afin de clarifier les causes des oestrogènes fœtaux élevés dans l'autisme.
Dans le cerveau, la signalisation médiée par les œstrogènes sur les neurones GABAergiques de l'hypothalamus est nécessaire pour supprimer l'axe stéroïdogène [51]. La suppression inefficace de cet axe dans l'autisme pourrait être due à une aromatisation inefficace des androgènes dans l'hypothalamus, à une résistance à la signalisation des œstrogènes et / ou à un dysfonctionnement du système GABAergique. Avant la naissance, la génétique fœtale (due par exemple à des mutations d'aromatase [52] ou d'activateurs d'aromatase [53]), de complications de la grossesse (par exemple, la taille du placenta [46]), ainsi que les facteurs de risque maternels (par exemple,PCOS [18]) pouvaient affecter divers points dans cette voie physiopathologique. Ces spéculations nécessiteraient des tests supplémentaires. Spécifiquement pour l'aromatisation, les ratios basés sur les taux d'hormones circulantes peuvent ne pas être suffisants pour capturer une activité spécifique du tissu, car l'aromatase est régulée de manière différenciée par des promoteurs séparés dans le placenta, les glandes surrénales et le cerveau [54].
Des taux élevés d'œstrogènes prénatals pourraient perturber de nombreux aspects de l'endocrinologie prénatale et affecter le développement prénatal du cerveau dans des zones qui ne se limitent pas à la différenciation sexuelle. Plusieurs sources de données plaident en faveur d’un rôle plus large de l’œstradiol en tant que «neurostéroïde» doté de nombreuses propriétés régulatrices [55]. Par exemple, la perturbation de la signalisation des œstrogènes dans le cervelet en développement des souris réduit la croissance des cellules de Purkinje chez les mâles et les femelles, mais ne réduit que le comportement social chez les souris mâles, ce qui suggère que le cervelet peut réagir à une perturbation œstrogénique de manière sexuellement dimorphique [56]. Au début du développement, l'œstradiol diminue la signalisation GABAergique [57] et facilite le passage postnatal de l'excitation à l'inhibition [28, 58].
Des taux élevés d'œstrogènes prénatals pourraient perturber de nombreux aspects de l'endocrinologie prénatale et affecter le développement prénatal du cerveau dans des zones qui ne se limitent pas à la différenciation sexuelle. Plusieurs sources de données plaident en faveur d’un rôle plus large de l’œstradiol en tant que «neurostéroïde» doté de nombreuses propriétés régulatrices [55]. Par exemple, la perturbation de la signalisation des œstrogènes dans le cervelet en développement des souris réduit la croissance des cellules de Purkinje chez les mâles et les femelles, mais ne réduit que le comportement social chez les souris mâles, ce qui suggère que le cervelet peut réagir à une perturbation œstrogénique de manière sexuellement dimorphique [56]. Au début du développement, l'œstradiol diminue la signalisation GABAergique [57] et facilite le passage postnatal de l'excitation à l'inhibition [28, 58].
Les œstrogènes augmentent à la fois le nombre d'épines sur les neurones corticaux primaires embryonnaires [55] et induisent le recrutement des protéines nécessaires à la formation de synapses excitatrices, telles que la neuroligine-1, la sous-unité NMDA GluN1 et la protéine de densité post-synaptique 95 (PSD-95) des épines [59]. Des niveaux plus élevés d'oestrogènes prénataux pourraient donc augmenter le nombre de synapses excitatrices dans le cortex, augmentant ainsi le risque d'autisme, comme le suggère la théorie excitation/ inhibition (E – I) de l'autisme [60]. Le phénotype perceptuel dans l'autisme est caractérisé par une inhibition réduite de GABAergic, comme le montrent des paradigmes tels que la rivalité binoculaire [61] et l'attention portée aux détails [62]. La signalisation par les œstrogènes pourrait donc être un modulateur significatif de l’inhibition neuronale, en particulier au début du développement du cerveau et de la «période critique» de la plasticité corticale, qui repose fortement sur le système GABAergique [63].
Bien que l'œstradiol (aromatisé à partir de la testostérone) soit le principal agent de masculinisation prénatale chez la plupart des mammifères [24], son rôle dans la différenciation sexuelle chez l'homme reste flou. Les hommes déficients en aromatase ont un développement typique de leur tractus urogénital [64], mais peuvent avoir des déficiences cognitives, une poussée de croissance insuffisante et des caractéristiques sexuelles secondaires atypiques telles que les proportions du corps féminisé à l’âge adulte [65]. Les œstrogènes peuvent donc féminiser et masculiniser les humains, en fonction du tissu cible et du milieu de développement. Dans l'autisme, les styles cognitifs et la neuroanatomie sexuellement dimorphique présentent des phénotypes masculinisés [7, 9, 10], mais la connectivité fonctionnelle et la croissance physique montrent un schéma mixte de changements masculins et féminins [8, 66]. Mais avant la naissance, et particulièrement pendant la période de masculinisation, le processus de différenciation sexuelle est censé être directionnel masculin par rapport à un mode par défaut anatomiquement et physiologiquement féminin. Les taux élevés d'œstrogènes fœtaux observés pourraient ainsi contribuer aux différences cognitives développementales [10], selon la théorie de l'autisme «du cerveau masculin extrême».
Il n’existait aucune association logistique univariée et statistiquement significative entre l’autisme et la testostérone ou l’androstènedione, qui agit via le récepteur des androgènes. Les mécanismes par lesquels la signalisation androgénique pourrait augmenter la probabilité d'autisme, qui ont peut-être été oubliés dans cette analyse, incluent des androgènes supplémentaires ou d'autres agonistes du récepteur des androgènes (par exemple, des neurostéroïdes tels que la déshydroépiandrostérone [67]), des effets d'interaction entre les androgènes et les œstrogènes (par exemple, la coactivation de la récepteur aux androgènes de l'œstradiol [68]), ainsi que des associations non linéaires d'androgènes à la probabilité d'autisme. Par conséquent, l'activité androgène peut encore constituer une caractéristique importante du développement de l'autisme, comme le suggèrent des comorbidités cliniques connexes [18, 69] et démontrée dans les associations de testostérone fœtale à des traits autistiques dans une cohorte séparée [70].
Nous n'avons pas pu déterminer si les œstrogènes prénatals étaient associés à la probabilité d'autisme chez les femmes, car il y avait trop peu de femmes diagnostiquées dans la CBH pendant cette période. Nous prévoyons de tester cela en élargissant la fenêtre temporelle. Ainsi, à l'heure actuelle, nos résultats ne concernent que les hommes. En outre, la comparaison des concentrations d'androgènes et de cortisol avec des œstrogènes est potentiellement confondue par le fait que ces derniers ont été analysés à une date ultérieure et ont subi un cycle supplémentaire de gel-dégel. Nous avons tenté de minimiser les sources potentielles de confusion en utilisant la même méthodologie de test avec l'analyse initiale (LC-MS / MS), ainsi que de réévaluer les différences de temps de stockage total dans ce sous-ensemble de la cohorte initiale (Tableau 1). 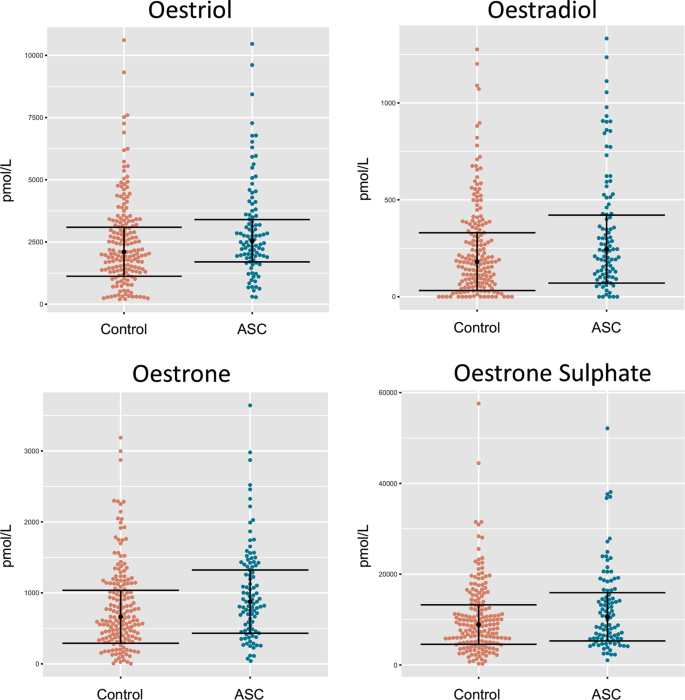
Une autre limite de cette étude est sa dépendance aux diagnostics cliniques du registre central des psychiatres du Danemark, que nous n'avons pas pu valider de manière indépendante. Cependant, une étude de validation antérieure des diagnostics d'autisme infantile figurant dans le registre central des psychiatres du Danemark a révélé que 94% des diagnostics enregistrés entre 1990 et 1999 étaient valides selon un schéma de codage normalisé [71]. De même, nous ne pouvons être certains de la source des stéroïdes amniotiques, car ils pourraient être d'origine fœtale, maternelle ou placentaire. Le plasma et le liquide amniotiques fœtaux sont en équilibre osmotique jusqu'à ce que la peau du fœtus soit kératinisée (généralement à 25 semaines de gestation) [72]. Par conséquent, les concentrations de stéroïdes dans le liquide amniotique reflètent avec précision celles de la circulation fœtale.
En conclusion, nous avons démontré que l'œstradiol, l'oestriol et l'œstrone prénataux sont élevés chez les garçons qui ont développé l'autisme. Cela étend notre constatation antérieure de stéroïdogenèse prénatale élevée dans la même cohorte et fournit des preuves supplémentaires de la théorie prénatale des stéroïdes de l'autisme [15]. Les taux élevés d'œstradiol prénatal contribuent davantage à la probabilité d'autisme que d'autres stéroïdes sexuels prénatals, y compris la testostérone. Nous concluons que l'excès œstrogénique prénatal est une caractéristique de l'autisme et qu'il peut interagir avec la prédisposition génétique à affecter le développement neurologique.
En conclusion, nous avons démontré que l'œstradiol, l'oestriol et l'œstrone prénataux sont élevés chez les garçons qui ont développé l'autisme. Cela étend notre constatation antérieure de stéroïdogenèse prénatale élevée dans la même cohorte et fournit des preuves supplémentaires de la théorie prénatale des stéroïdes de l'autisme [15]. Les taux élevés d'œstradiol prénatal contribuent davantage à la probabilité d'autisme que d'autres stéroïdes sexuels prénatals, y compris la testostérone. Nous concluons que l'excès œstrogénique prénatal est une caractéristique de l'autisme et qu'il peut interagir avec la prédisposition génétique à affecter le développement neurologique.
Foetal oestrogens and autism
Molecular Psychiatry
(2019)
Abstract
Elevated
latent prenatal steroidogenic activity has been found in the amniotic
fluid of autistic boys, based on measuring prenatal androgens and other
steroid hormones. To date, it is unclear if other prenatal steroids also
contribute to autism likelihood. Prenatal oestrogens need to be
investigated, as they play a key role in synaptogenesis and
corticogenesis during prenatal development, in both males and females.
Here we test whether levels of prenatal oestriol, oestradiol, oestrone
and oestrone sulphate in amniotic fluid are associated with autism, in
the same Danish Historic Birth Cohort, in which prenatal androgens were
measured, using univariate logistic regression (n = 98 cases, n = 177
controls). We also make a like-to-like comparison between the prenatal
oestrogens and androgens. Oestradiol, oestrone, oestriol and
progesterone each related to autism in univariate analyses after
correction with false discovery rate. A comparison of standardised odds
ratios showed that oestradiol, oestrone and progesterone had the largest
effects on autism likelihood. These results for the first time show
that prenatal oestrogens contribute to autism likelihood, extending the
finding of elevated prenatal steroidogenic activity in autism. This
likely affects sexual differentiation, brain development and function.
Introduction
The male-biased prevalence of autism [1, 2], together with the finding that autistic girls have a higher mutational load than autistic boys [3,4,5],
suggests that males have a higher likelihood of developing autism. The
sex ratio in autism diagnoses persists even after taking into account
under- and/or mis-diagnosis, as well as camouflaging in females, with
males being three times more likely to have the condition [6].
This implicates mechanisms of sexual differentiation in the development
of autism. Five recent findings support this inference.
First, autistic women have atypical brain structure in sexually dimorphic regions, when assessed via magnetic resonance imaging and compared to neurotypical controls [7]. Second, functional connectivity in the brain of males with autism shows both hypermasculine and hyperfeminine patterns, when assessed in relation to neurotypical sex differences [8]. Third, autistic people show a masculinised shift in scores on two key sexually dimorphic psychological traits, empathy and systemising, a finding that has been replicated in a big data study of 36,000 autistic people [9, 10]. Fourth, autistic women have elevated androstenedione levels, the precursor to testosterone [11]. Finally, fifth, autistic children have hypermasculine facial features, as rated using three-dimensional photogrammetry [12].
Although autism is strongly heritable and sex-associated genetic mechanisms could contribute to this implication of sexual differentiation in autism [2, 5], prenatal hormone exposure and a brief surge in foetal testosterone are critical for sexual differentiation and masculinisation in humans [13, 14]. In line with this, we previously found elevated steroidogenic activity during this prenatal masculinisation window (PMW) in the amniotic fluid of autistic boys [15]. Subsequently, three very large epidemiological studies revealed a link between autism and maternal polycystic ovarian syndrome (PCOS), a condition associated with androgenic excess [16,17,18]. Consistently with this, the 2D:4D digit ratio, a marker of prenatal androgen exposure, is also masculinised in autistic children and their parents [19]. Finally, autistic women and their mothers have elevated rates of steroid-related cancers, such as breast cancer and ovarian cancer [20].
However, a number of studies that focused on testosterone have not replicated the correlation of hormonal levels with autistic traits. First, umbilical cord testosterone measured soon after birth was not associated with the development of autistic traits [21]. Second, salivary testosterone during a brief period of postnatal steroid surge (‘mini-puberty’) also did not correlate with autistic traits in toddlers [22]. In both cases, testosterone was measured postnatally—in the neonatal period—rather than during the PMW, during which foetal testosterone is first produced and masculinisation of the brain and body commences. This would suggest that timing is critical for the effects of testosterone on the brain, with the late first-early second trimester PMW being key, rather than the neonatal period. Finally, univariate assessment of amniotic testosterone in a separate cohort of neurotypical children also failed to reveal an association to autistic traits in childhood [23]. This latter finding may reflect that the wider endocrine environment outside testosterone is also significant for autism likelihood.
While prenatal androgens are responsible for masculinisation in humans, prenatal oestrogens also contribute to foetal and neonatal brain development [24], and yet these have not been thoroughly investigated for their potential role in autism likelihood. Oestrogens and their receptors are widespread in the developing brain in both males and females and regulate many neurodevelopmental processes, including synaptogenesis, apoptosis and neuronal differentiation [25,26,27]. Oestradiol in particular supports synapse formation in the cortex by enhancing excitatory GABA activity [28]. In autism, synapse formation [29], neuronal differentiation [30] as well as the GABAergic system [31] are all atypical. These provide clues that prenatal oestrogens may be involved in autism. However, we still lack direct evidence of this.
With regard to clinical studies in humans, low oestriol in maternal serum during the second trimester of pregnancy significantly increases the likelihood of autism in the foetus, as demonstrated in a large study of n = 2586 autistic pregnancies [32]. This study may have been confounded by a variety of pregnancy complications, such as pre-eclampsia [33] and being small for gestational age [34], since these are also more frequent in autism [35,36,37]. Thus, further study of prenatal oestrogenic activity, particularly in foetal circulation, is warranted. In addition, there is a need to compare different prenatal oestrogens to each other, in relation to autism likelihood.
In the present study, we measured prenatal levels of oestriol, oestradiol, oestrone and oestrone sulphate in amniotic fluid of boys with and without autism (n = 98 and n = 177 respectively) from the Danish Historic Birth Cohort (HBC), in the same samples in which we had found an elevated steroidogenic factor, following principal component analysis of prenatal androgens and other steroid hormones [15]. We have expanded on these findings by assaying oestrogens and by assessments of each steroid hormone to autism likelihood via univariate logistic regression. We also investigated potential differences in the aromatising capacity in autism by comparing the ratio between androgens and oestrogens. Finally, we calculated standardised effect sizes for all hormones assayed to date in this cohort, in order to understand which amniotic fluid hormones make the largest contribution to autism likelihood.
First, autistic women have atypical brain structure in sexually dimorphic regions, when assessed via magnetic resonance imaging and compared to neurotypical controls [7]. Second, functional connectivity in the brain of males with autism shows both hypermasculine and hyperfeminine patterns, when assessed in relation to neurotypical sex differences [8]. Third, autistic people show a masculinised shift in scores on two key sexually dimorphic psychological traits, empathy and systemising, a finding that has been replicated in a big data study of 36,000 autistic people [9, 10]. Fourth, autistic women have elevated androstenedione levels, the precursor to testosterone [11]. Finally, fifth, autistic children have hypermasculine facial features, as rated using three-dimensional photogrammetry [12].
Although autism is strongly heritable and sex-associated genetic mechanisms could contribute to this implication of sexual differentiation in autism [2, 5], prenatal hormone exposure and a brief surge in foetal testosterone are critical for sexual differentiation and masculinisation in humans [13, 14]. In line with this, we previously found elevated steroidogenic activity during this prenatal masculinisation window (PMW) in the amniotic fluid of autistic boys [15]. Subsequently, three very large epidemiological studies revealed a link between autism and maternal polycystic ovarian syndrome (PCOS), a condition associated with androgenic excess [16,17,18]. Consistently with this, the 2D:4D digit ratio, a marker of prenatal androgen exposure, is also masculinised in autistic children and their parents [19]. Finally, autistic women and their mothers have elevated rates of steroid-related cancers, such as breast cancer and ovarian cancer [20].
However, a number of studies that focused on testosterone have not replicated the correlation of hormonal levels with autistic traits. First, umbilical cord testosterone measured soon after birth was not associated with the development of autistic traits [21]. Second, salivary testosterone during a brief period of postnatal steroid surge (‘mini-puberty’) also did not correlate with autistic traits in toddlers [22]. In both cases, testosterone was measured postnatally—in the neonatal period—rather than during the PMW, during which foetal testosterone is first produced and masculinisation of the brain and body commences. This would suggest that timing is critical for the effects of testosterone on the brain, with the late first-early second trimester PMW being key, rather than the neonatal period. Finally, univariate assessment of amniotic testosterone in a separate cohort of neurotypical children also failed to reveal an association to autistic traits in childhood [23]. This latter finding may reflect that the wider endocrine environment outside testosterone is also significant for autism likelihood.
While prenatal androgens are responsible for masculinisation in humans, prenatal oestrogens also contribute to foetal and neonatal brain development [24], and yet these have not been thoroughly investigated for their potential role in autism likelihood. Oestrogens and their receptors are widespread in the developing brain in both males and females and regulate many neurodevelopmental processes, including synaptogenesis, apoptosis and neuronal differentiation [25,26,27]. Oestradiol in particular supports synapse formation in the cortex by enhancing excitatory GABA activity [28]. In autism, synapse formation [29], neuronal differentiation [30] as well as the GABAergic system [31] are all atypical. These provide clues that prenatal oestrogens may be involved in autism. However, we still lack direct evidence of this.
With regard to clinical studies in humans, low oestriol in maternal serum during the second trimester of pregnancy significantly increases the likelihood of autism in the foetus, as demonstrated in a large study of n = 2586 autistic pregnancies [32]. This study may have been confounded by a variety of pregnancy complications, such as pre-eclampsia [33] and being small for gestational age [34], since these are also more frequent in autism [35,36,37]. Thus, further study of prenatal oestrogenic activity, particularly in foetal circulation, is warranted. In addition, there is a need to compare different prenatal oestrogens to each other, in relation to autism likelihood.
In the present study, we measured prenatal levels of oestriol, oestradiol, oestrone and oestrone sulphate in amniotic fluid of boys with and without autism (n = 98 and n = 177 respectively) from the Danish Historic Birth Cohort (HBC), in the same samples in which we had found an elevated steroidogenic factor, following principal component analysis of prenatal androgens and other steroid hormones [15]. We have expanded on these findings by assaying oestrogens and by assessments of each steroid hormone to autism likelihood via univariate logistic regression. We also investigated potential differences in the aromatising capacity in autism by comparing the ratio between androgens and oestrogens. Finally, we calculated standardised effect sizes for all hormones assayed to date in this cohort, in order to understand which amniotic fluid hormones make the largest contribution to autism likelihood.
Discussion
This
study reports the first evidence that elevated levels of prenatal
amniotic oestradiol, oestriol and oestrone are each associated with
autism, with oestradiol levels being the most significant predictor of
autism likelihood in univariate logistic regression models. These
findings complement earlier observations that elevated steroidogenic
activity is associated with autism in the same samples derived from the
Danish Historic Birth cohort [15].
We also calculated standardised ORs, in order to directly compare the
effect sizes of all amniotic steroids measured to date. We found that
oestradiol had the strongest positive effect size on autism likelihood,
followed by oestrone, oestriol and progesterone (Fig. 3). This finding appears to contradict an earlier report by Windham et al. [32] that showed that lower
levels of oestriol in second trimester were modestly associated with a
later diagnosis of autism in the offspring. However, our samples
correspond to a slightly earlier time point in pregnancy compared to
Windham et al. (mean gestational week = 14.9 vs. 17.2 respectively) (see
Table 1) [32], which could potentially better capture the steroid surge during the PMW [14].
Furthermore, our samples are of different origin, as Windham et al.
assayed maternal serum, rather than foetal amniotic fluid. Steroid
hormone levels in maternal serum do not differ relative to the baby’s
sex and do not correlate to amniotic levels during the PMW [42].
Therefore, amniotic oestrogens are arguably more relevant to the
current research question than are maternal serum oestrogens.
A discrepancy in oestrogen levels between the mother and child could potentially be attributed to the placenta, which acts as an endocrine regulator of the maternal–foetal interface and the main source of oestrogen production for the foetus via the aromatisation of androgens [43]. Several lines of evidence suggest a contributory role for the placenta in the aetiology of autism. First, there is increased placental inflammation in autism [44]. Second, there is atypical placental morphology [45] and increased placental size [46] in cases of autism and at high familial risk respectively. Third, complications related to the placenta (pre-eclampsia [47], hypertensive disorders [48]) are also more frequent in pregnancies that lead to autism. As with autism, placental dysfunction also disproportionately affects males more than females [49].
Given the high pairwise correlations between many of the steroid hormones (Fig. 2, Supplementary Table 3), as well as a lack of difference in aromatisation between cases and controls, our data suggest that an increase in foetal oestrogens is secondary to increased activity along the entirety of the steroidogenic axis in pregnancies that later result in autism [15]. Interestingly, oestradiol was not significantly correlated to testosterone (Pearson’s β = 0.007, p = 0.9103) despite their proximity in steroidogenesis. This discrepancy may be because oestrogens are also de novo produced by the placenta, in addition to being aromatised from foetal and maternal androgens [43, 50]. Thus, a multi-systems approach is needed in order to clarify the causes of elevated foetal oestrogens in autism.
In the brain, oestrogen-mediated signalling on GABAergic neurons in the hypothalamus is required in order to suppress the steroidogenic axis [51]. Inefficient suppression of this axis in autism could be due to inefficient aromatisation of androgens in the hypothalamus, resistance to oestrogen signalling and/or dysfunction of the GABAergic system. Prenatally, foetal genetics (e.g. due to mutations in aromatase [52] or aromatase activators [53]), pregnancy complications (e.g. placental size [46]), as well as maternal risk factors (e.g. PCOS [18]) could all affect various points in this pathophysiologic pathway. These speculations would require further testing. Specifically for aromatisation, ratios based on circulating hormone levels may not be sufficient to capture tissue-specific activity, since aromatase is differentially regulated by separate promoters in the placenta, the adrenals and the brain [54].
High levels of prenatal oestrogens could dysregulate many aspects of prenatal endocrinology and affect prenatal brain development in areas that are not restricted to sexual differentiation. Several lines of evidence support a wider role of oestradiol as a ‘neurosteroid’ with many regulatory properties [55]. For example, disruption of oestrogen signalling in the developing cerebellum of mice reduces Purkinje cell growth in both males and females, but only reduces social behaviour in male mice, suggesting that the cerebellum may react to oestrogenic disruption in a sexually dimorphic way [56]. In early development, oestradiol decreases GABAergic signalling [57] and mediates its postnatal shift from excitation to inhibition [28, 58]. Oestrogens both increase the number of spines on embryonic primary cortical neurons [55] and induce the recruitment of proteins necessary for excitatory synapse formation, such as neuroligin-1, NMDA subunit GluN1, and post-synaptic density protein 95 (PSD-95) to the spines [59]. Higher levels of prenatal oestrogens might therefore increase the number of excitatory synapses in the cortex, increasing the likelihood for autism, as suggested by the excitatory/inhibitory (E–I) theory of autism [60]. The perceptual phenotype in autism is characterised by reduced GABAergic inhibition, as shown using paradigms such as binocular rivalry [61] and attention to detail [62]. Oestrogen signalling could thus be a significant modulator of neuronal inhibition, particularly during early brain development and the ‘critical period’ of cortical plasticity, which is heavily reliant on the GABAergic system [63].
Although oestradiol (aromatised from testosterone) is the main prenatal masculinising agent in most mammals [24], its role in human sexual differentiation remains unclear. Men with aromatase deficiency have typical development of their urogenital tract [64], but may have cognitive disabilities, lack a growth spurt, and have atypical secondary sexual characteristics such as feminised body proportions in adulthood [65]. Oestrogens may therefore both feminise and masculinise humans, depending on the target tissue and developmental milieu. In autism, cognitive styles and sexually dimorphic neuroanatomy show some masculinised phenotypes [7, 9, 10], but functional connectivity and physical growth show a mixed pattern of masculine and feminine shifts [8, 66]. Prenatally though, and particularly during the masculinisation window, the process of sexual differentiation is understood to be directionally masculine over an anatomically and physiologically female default. The observed high levels of foetal oestrogens could thus be contributing to developmental cognitive differences [10], according to the “extreme male brain” theory of autism.
There was no statistically significant univariate, logistic association between autism and testosterone or androstenedione, which act via the androgen receptor. Mechanisms through which androgenic signalling could increase autism likelihood, which may have been missed in this analysis, include additional androgens or other agonists of the androgen receptor (e.g. neurosteroids like dehydroepiandrosterone [67]), interaction effects between androgens and oestrogens (e.g. coactivation of the androgen receptor by oestradiol [68]), as well as non-linear associations of androgens to autism likelihood. Consequently, androgenic activity may still be an important feature in the development of autism, as suggested by related clinical comorbidities [18, 69] and shown in associations of foetal testosterone to autistic traits in a separate cohort [70].
We could not test whether prenatal oestrogens were associated with autism likelihood in females as there were too few diagnosed women in the HBC in this time window. We plan to test this by expanding the time window. Thus, at present, our findings only generalise to males. Furthermore, comparison of the concentrations of androgens and cortisol to oestrogens is potentially confounded by the fact that the latter were analysed at a later time point and underwent an additional freeze–thaw cycle. We have attempted to minimise any potential sources of confounding by using the same assay methodology with the initial analysis (LC-MS/MS), as well as reassessing for any differences in total storage time in this subset of the original cohort (Table 1).
Another limitation of this study is its reliance on clinical diagnoses from the Danish Central Psychiatric Register, which we could not be independently validated. However, a previous validation study of childhood autism diagnoses in the Danish Central Psychiatric Register found that 94% of diagnoses between 1990 and 1999 in the register were valid using a standardised coding scheme [71]. Similarly, we cannot be certain about the source of amniotic steroids, as they could be of foetal, maternal or placental origin. Foetal plasma and amniotic fluid are in osmotic equilibrium until the foetal skin is keratinised (typically by 25 weeks of gestation) [72]. Therefore, steroid concentrations in amniotic fluid accurately reflect those in foetal circulation.
In conclusion, we have demonstrated that prenatal oestradiol, oestriol and oestrone are elevated in in boys who went on to develop autism. This extends our previous finding of elevated prenatal steroidogenesis in the same cohort and provides further evidence for the prenatal steroid theory of autism [15]. High levels of prenatal oestradiol contribute to a greater degree to autism likelihood than other prenatal sex steroids, including testosterone. We conclude that prenatal oestrogenic excess is a characteristic of autism and may interact with genetic predisposition to affect neurodevelopment.
A discrepancy in oestrogen levels between the mother and child could potentially be attributed to the placenta, which acts as an endocrine regulator of the maternal–foetal interface and the main source of oestrogen production for the foetus via the aromatisation of androgens [43]. Several lines of evidence suggest a contributory role for the placenta in the aetiology of autism. First, there is increased placental inflammation in autism [44]. Second, there is atypical placental morphology [45] and increased placental size [46] in cases of autism and at high familial risk respectively. Third, complications related to the placenta (pre-eclampsia [47], hypertensive disorders [48]) are also more frequent in pregnancies that lead to autism. As with autism, placental dysfunction also disproportionately affects males more than females [49].
Given the high pairwise correlations between many of the steroid hormones (Fig. 2, Supplementary Table 3), as well as a lack of difference in aromatisation between cases and controls, our data suggest that an increase in foetal oestrogens is secondary to increased activity along the entirety of the steroidogenic axis in pregnancies that later result in autism [15]. Interestingly, oestradiol was not significantly correlated to testosterone (Pearson’s β = 0.007, p = 0.9103) despite their proximity in steroidogenesis. This discrepancy may be because oestrogens are also de novo produced by the placenta, in addition to being aromatised from foetal and maternal androgens [43, 50]. Thus, a multi-systems approach is needed in order to clarify the causes of elevated foetal oestrogens in autism.
In the brain, oestrogen-mediated signalling on GABAergic neurons in the hypothalamus is required in order to suppress the steroidogenic axis [51]. Inefficient suppression of this axis in autism could be due to inefficient aromatisation of androgens in the hypothalamus, resistance to oestrogen signalling and/or dysfunction of the GABAergic system. Prenatally, foetal genetics (e.g. due to mutations in aromatase [52] or aromatase activators [53]), pregnancy complications (e.g. placental size [46]), as well as maternal risk factors (e.g. PCOS [18]) could all affect various points in this pathophysiologic pathway. These speculations would require further testing. Specifically for aromatisation, ratios based on circulating hormone levels may not be sufficient to capture tissue-specific activity, since aromatase is differentially regulated by separate promoters in the placenta, the adrenals and the brain [54].
High levels of prenatal oestrogens could dysregulate many aspects of prenatal endocrinology and affect prenatal brain development in areas that are not restricted to sexual differentiation. Several lines of evidence support a wider role of oestradiol as a ‘neurosteroid’ with many regulatory properties [55]. For example, disruption of oestrogen signalling in the developing cerebellum of mice reduces Purkinje cell growth in both males and females, but only reduces social behaviour in male mice, suggesting that the cerebellum may react to oestrogenic disruption in a sexually dimorphic way [56]. In early development, oestradiol decreases GABAergic signalling [57] and mediates its postnatal shift from excitation to inhibition [28, 58]. Oestrogens both increase the number of spines on embryonic primary cortical neurons [55] and induce the recruitment of proteins necessary for excitatory synapse formation, such as neuroligin-1, NMDA subunit GluN1, and post-synaptic density protein 95 (PSD-95) to the spines [59]. Higher levels of prenatal oestrogens might therefore increase the number of excitatory synapses in the cortex, increasing the likelihood for autism, as suggested by the excitatory/inhibitory (E–I) theory of autism [60]. The perceptual phenotype in autism is characterised by reduced GABAergic inhibition, as shown using paradigms such as binocular rivalry [61] and attention to detail [62]. Oestrogen signalling could thus be a significant modulator of neuronal inhibition, particularly during early brain development and the ‘critical period’ of cortical plasticity, which is heavily reliant on the GABAergic system [63].
Although oestradiol (aromatised from testosterone) is the main prenatal masculinising agent in most mammals [24], its role in human sexual differentiation remains unclear. Men with aromatase deficiency have typical development of their urogenital tract [64], but may have cognitive disabilities, lack a growth spurt, and have atypical secondary sexual characteristics such as feminised body proportions in adulthood [65]. Oestrogens may therefore both feminise and masculinise humans, depending on the target tissue and developmental milieu. In autism, cognitive styles and sexually dimorphic neuroanatomy show some masculinised phenotypes [7, 9, 10], but functional connectivity and physical growth show a mixed pattern of masculine and feminine shifts [8, 66]. Prenatally though, and particularly during the masculinisation window, the process of sexual differentiation is understood to be directionally masculine over an anatomically and physiologically female default. The observed high levels of foetal oestrogens could thus be contributing to developmental cognitive differences [10], according to the “extreme male brain” theory of autism.
There was no statistically significant univariate, logistic association between autism and testosterone or androstenedione, which act via the androgen receptor. Mechanisms through which androgenic signalling could increase autism likelihood, which may have been missed in this analysis, include additional androgens or other agonists of the androgen receptor (e.g. neurosteroids like dehydroepiandrosterone [67]), interaction effects between androgens and oestrogens (e.g. coactivation of the androgen receptor by oestradiol [68]), as well as non-linear associations of androgens to autism likelihood. Consequently, androgenic activity may still be an important feature in the development of autism, as suggested by related clinical comorbidities [18, 69] and shown in associations of foetal testosterone to autistic traits in a separate cohort [70].
We could not test whether prenatal oestrogens were associated with autism likelihood in females as there were too few diagnosed women in the HBC in this time window. We plan to test this by expanding the time window. Thus, at present, our findings only generalise to males. Furthermore, comparison of the concentrations of androgens and cortisol to oestrogens is potentially confounded by the fact that the latter were analysed at a later time point and underwent an additional freeze–thaw cycle. We have attempted to minimise any potential sources of confounding by using the same assay methodology with the initial analysis (LC-MS/MS), as well as reassessing for any differences in total storage time in this subset of the original cohort (Table 1).
Another limitation of this study is its reliance on clinical diagnoses from the Danish Central Psychiatric Register, which we could not be independently validated. However, a previous validation study of childhood autism diagnoses in the Danish Central Psychiatric Register found that 94% of diagnoses between 1990 and 1999 in the register were valid using a standardised coding scheme [71]. Similarly, we cannot be certain about the source of amniotic steroids, as they could be of foetal, maternal or placental origin. Foetal plasma and amniotic fluid are in osmotic equilibrium until the foetal skin is keratinised (typically by 25 weeks of gestation) [72]. Therefore, steroid concentrations in amniotic fluid accurately reflect those in foetal circulation.
In conclusion, we have demonstrated that prenatal oestradiol, oestriol and oestrone are elevated in in boys who went on to develop autism. This extends our previous finding of elevated prenatal steroidogenesis in the same cohort and provides further evidence for the prenatal steroid theory of autism [15]. High levels of prenatal oestradiol contribute to a greater degree to autism likelihood than other prenatal sex steroids, including testosterone. We conclude that prenatal oestrogenic excess is a characteristic of autism and may interact with genetic predisposition to affect neurodevelopment.
References
- 1.Fombonne E. Epidemiology of pervasive developmental disorders. Pediatr Res. 2009;65:591–8.
- 2.Baron-Cohen S, Lombardo MV, Auyeung B, Ashwin E, Chakrabarti B, Knickmeyer R. Why are autism spectrum conditions more prevalent in males? PLoS Biol. 2011;9:e1001081.
- 3.Robinson EB, Lichtenstein P, Anckarsäter H, Happé F, Ronald A. Examining and interpreting the female protective effect against autistic behavior. Proc Natl Acad Sci USA. 2013;110:5258–62.
- 4.Jacquemont S, Coe BP, Hersch M, Duyzend MH, Krumm N, Bergmann S, et al. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am J Hum Genet. 2014;94:415–25.
- 5.Werling DM, Geschwind DH. Recurrence rates provide evidence for sex-differential, familial genetic liability for autism spectrum disorders in multiplex families and twins. Mol Autism. 2015;6:1–14.
- 6.Loomes R, Hull L, Mandy WPL. What is the male-to-female ratio in autism spectrum disorder? a systematic review and meta-analysis. J Am Acad Child Adolesc Psychiatry]. 2017;56:466–74.
- 7.Lai MC, Lombardo MV, Suckling J, Ruigrok AN, Chakrabarti B, Ecker C, et al. Biological sex affects the neurobiology of autism. Brain. 2013;136(Pt 9):2799–815.
- 8.Floris DL, Lai MC, Nath T, Milham MP, Di Martino A. Network-specific sex differentiation of intrinsic brain function in males with autism. Mol Autism. 2018;9:17.
- 9.Baron-Cohen S, Cassidy S, Auyeung B, Allison C, Achoukhi M, Robertson S, et al. Attenuation of typical sex differences in 800 adults with autism vs. 3,900 controls. PLoS ONE. 2014;9:e102251.
- 10.Greenberg DM, Warrier V, Allison C, Baron-Cohen S. Testing the empathizing-systemizing theory of sex differences and the extreme male brain theory of autism in half a million people. Proc Natl Acad Sci USA. 2018;115:12152–7.
- 11.Schwarz E, Guest PC, Rahmoune H, Wang L, Levin Y, Ingudomnukul E, et al. Sex-specific serum biomarker patterns in adults with Asperger’s syndrome. Mol Psychiatry. 2011;16:1213–20.
- 12.Tan DW, Gilani SZ, Maybery MT, Mian A, Hunt A, Walters M, et al. Hypermasculinised facial morphology in boys and girls with Autism Spectrum Disorder and its association with symptomatology. Sci Rep. 2017;7:9348.
- 13.Hines M, Constantinescu M, Spencer D. Early androgen exposure and human gender development. Biol Sex Differ. 2015;6:3.
- 14.Welsh M, Suzuki H, Yamada G. The Masculinization Programming Window. Endocr Dev. 2014;27:17–27.
- 15.Baron-Cohen S, Auyeung B, Nørgaard-Pedersen B, Hougaard DM, Abdallah MW, Melgaard L, et al. Elevated fetal steroidogenic activity in autism. Mol Psychiatry. 2015;20:369–76.
- 16.Kosidou K, Dalman C, Widman L, Arver S, Lee BK, Magnusson C, et al. Maternal polycystic ovary syndrome and the risk of autism spectrum disorders in the offspring: a population-based nationwide study in Sweden. Mol Psychiatry. 2016;21:1441–8.
- 17.Berni TR, Morgan CL, Berni ER, Rees DA. Polycystic ovary syndrome is associated with adverse mental health and neurodevelopmental outcomes. J Clin Endocrinol Metab. 2018;103:2116–25.
- 18.Cherskov A, Pohl A, Allison C, Zhang H, Payne RA, Baron-Cohen S. Polycystic ovary syndrome and autism: a test of the prenatal sex steroid theory. Transl Psychiatry. 2018;8:136.
- 19.Manning JT, Baron-Cohen S, Wheelwright S, Sanders G. The 2nd to 4th digit ratio and autism. Dev Med Child Neurol. 2001;43:160–4.
- 20.Ingudomnukul E, Baron-Cohen S, Wheelwright S, Knickmeyer R. Elevated rates of testosterone-related disorders in women with autism spectrum conditions. Horm Behav]. 2007;51:597–604.
- 21.Whitehouse AJ, Mattes E, Maybery MT, Dissanayake C, Sawyer M, Jones RM, et al. Perinatal testosterone exposure and autistic-like traits in the general population: a longitudinal pregnancy-cohort study. J Neurodev Disord. 2012;4:25.
- 22.Kung KT, Constantinescu M, Browne WV, Noorderhaven RM, Hines M. No relationship between early postnatal testosterone concentrations and autistic traits in 18 to 30-month-old children. Mol Autism. 2016;7:15.
- 23.Kung KT, Spencer D, Pasterski V, Neufeld S, Glover V, O’Connor TG, et al. No relationship between prenatal androgen exposure and autistic traits: convergent evidence from studies of children with congenital adrenal hyperplasia and of amniotic testosterone concentrations in typically developing children. J Child Psychol Psychiatry. 2016;57:1455–62.
- 24.McCarthy MM. Estradiol and the developing brain. Physiol Rev. 2008;88:91–134.
- 25.Konkle ATM, McCarthy MM. Developmental time course of estradiol, testosterone, and dihydrotestosterone levels in discrete regions of male and female rat brain. Endocrinology 2011;152:223–35.
- 26.MacLusky NJ, Naftolin F. Sexual differentiation of the central nervous system. Science. 1981;211:1294–302.
- 27.González M, Cabrera-Socorro A, Pérez-García CG, Fraser JD, López FJ, Alonso R, et al. Distribution patterns of estrogen receptor alpha and beta in the human cortex and hippocampus during development and adulthood. J Comp Neurol. 2007;503:790–802.
- 28.Nunez JL, Aberdeen GW, Albrecht ED, McCarthy MM. Impact of estradiol on gamma-aminobutyric acid- and glutamate-mediated calcium responses of fetal baboon (Papio anubis) hippocampal and cortical neurons. Endocrinology 2008;149:6433–43.
- 29.Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry. 2012;17:71–84.
- 30.Li J, Shi M, Ma Z, Zhao S, Euskirchen G, Ziskin J, et al. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol Syst Biol. 2014;10:774.
- 31.Puts NAJ, Wodka EL, Harris AD, Crocetti D, Tommerdahl M, Mostofsky SH, et al. Reduced GABA and altered somatosensory function in children with autism spectrum disorder. Autism Res. 2017;10:608–19.
- 32.Windham GC, Lyall K, Anderson M, Kharrazi M. Autism spectrum disorder risk in relation to maternal mid-pregnancy serum hormone and protein markers from prenatal screening in California. J Autism Dev Disord. 2016;46:478–88.
- 33.Tache V, Baer RJ, Currier RJ, Li CS, Towner D, Waetjen LE, et al. Population-based biomarker screening and the development of severe preeclampsia in California. Am J Obstet Gynecol. 2014;211:377.e1–377.e8.
- 34.Talge NM, Holzman C, Senagore PK, Klebanoff M, Fisher R. Biological indicators of the in-utero environment and their association with birth weight for gestational age. J Dev Orig Health Dis. 2011;2:280–90.
- 35.Walker CK, Krakowiak P, Baker A, Hansen RL, Ozonoff S, Hertz-Picciotto I. Preeclampsia, placental insufficiency, and autism spectrum disorder or developmental delay. JAMA Pediatr. 2015;169:154.
- 36.Moore GS, Kneitel AW, Walker CK, Gilbert WM, Xing G. Autism risk in small- and large-for-gestational-age infants. Am J Obstet Gynecol. 2012;206:314.e1–9.
- 37.Lyall K, Pauls DL, Spiegelman D, Ascherio A, Santangelo SL. Pregnancy complications and obstetric suboptimality in association with autism spectrum disorders in children of the Nurses’ Health Study II. Autism Res. 2012;5:21–30.
- 38.Faul F, Erdfelder E, Buchner A, Lang A-G. Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav Res Methods. 2009;41:1149–60.
- 39.Weichman BM, Notides AC. Estrogen receptor activation and the dissociation kinetics of estradiol, estriol, and estrone. Endocrinology 1980;106:434–9.
- 40.Korenman SG. Comparative binding affinity of estrogens. Steroids 1968;13:163–77.
- 41.Sollberger S, Ehlert U. How to use and interpret hormone ratios. Psychoneuroendocrinology. 2016;63:385–97.
- 42.van de Beek C, Thijssen JH, Cohen-Kettenis PT, van Goozen SH, Buitelaar JK. Relationships between sex hormones assessed in amniotic fluid, and maternal and umbilical cord serum: what is the best source of information to investigate the effects of fetal hormonal exposure? Horm Behav. 2004;46:663–9.
- 43.Kaludjerovic J, Ward WE. The Interplay between Estrogen and Fetal Adrenal Cortex. J Nutr Metab. 2012;2012:837901.
- 44.Straughen JK, Misra DP, Divine G, Shah R, Perez G, VanHorn S, et al. The association between placental histopathology and autism spectrum disorder. Placenta. 2017;57:183–8.
- 45.Anderson GM, Jacobs-Stannard A, Chawarska K, Volkmar FR, Kliman HJ. Placental trophoblast inclusions in autism spectrum disorder. Biol Psychiatry. 2007;61:487–91.
- 46.Park BY, Misra DP, Moye J, Miller RK, Croen L, Fallin MD, et al. Placental gross shape differences in a high autism risk cohort and the general population. PLoS One. 2018;13:e0191276.
- 47.Dachew BA, Mamun A, Maravilla JC, Alati R. Pre-eclampsia and the risk of autism-spectrum disorder in offspring: meta-analysis. Br J Psychiatry. 2018;212:142–7.
- 48.Curran EA, O’Keeffe GW, Looney AM, Moloney G, Hegarty SV, Murray DM, et al. Exposure to hypertensive disorders of pregnancy increases the risk of autism spectrum disorder in affected offspring. Mol Neurobiol. 2018;55:5557–64.
- 49.Murji A, Proctor LK, Paterson AD, Chitayat D, Weksberg R, Kingdom J. Male sex bias in placental dysfunction. Am J Med Genet A. 2012;158A:779–83.
- 50.Escobar JC, Patel SS, Beshay VE, Suzuki T, Carr BR. The human placenta expresses CYP17 and generates androgens de novo. J Clin Endocrinol Metab. 2011;96:1385–92.
- 51.Pitteloud N, Dwyer AA, DeCruz S, Lee H, Boepple PA, Crowley WF Jr., et al. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypothalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab . 2008;93:784–91.
- 52.Chakrabarti B, Dudbridge F, Kent L, Wheelwright S, Hill-Cawthorne G, Allison C, et al. Genes related to sex steroids, neural growth, and social-emotional behavior are associated with autistic traits, empathy, and Asperger syndrome. Autism Res. 2009;2:157–77.
- 53.Sarachana T, Hu VW. Genome-wide identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol Autism. 2013;4:14.
- 54.Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Corbin CJ, Mendelson CR. Tissue-specific promoters regulate aromatase cytochrome P450 expression. Clin Chem. 1993;39:317–24.
- 55.Srivastava DP, Woolfrey KM, Liu F, Brandon NJ, Penzes P. Estrogen receptor beta activity modulates synaptic signaling and structure. J Neurosci. 2010;30:13454–60.
- 56.Hoffman JF, Wright CL, McCarthy MM. A critical period in purkinje cell development is mediated by local estradiol synthesis, disrupted by inflammation, and has enduring consequences only for males. J Neurosci. 2016;36:10039–49.
- 57.Mukherjee J, Cardarelli RA, Cantaut-Belarif Y, Deeb TZ, Srivastava DP, Tyagarajan SK, et al. Estradiol modulates the efficacy of synaptic inhibition by decreasing the dwell time of GABAA receptors at inhibitory synapses. Proc Natl Acad Sci USA. 2017;114:11763–8.
- 58.Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell. 2001;105:521–32.
- 59.Sellers KJ, Erli F, Raval P, Watson IA, Chen D, Srivastava DP. Rapid modulation of synaptogenesis and spinogenesis by 17β-estradiol in primary cortical neurons. Front Cell Neurosci. 2015;9:137.
- 60.Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–67.
- 61.Robertson CE, Kravitz DJ, Freyberg J, Baron-Cohen S, Baker CI. Slower rate of binocular rivalry in autism. J Neurosci. 2013;33:16983–91.
- 62.Robertson CE, Kravitz DJ, Freyberg J, Baron-Cohen S, Baker CI. Tunnel vision: sharper gradient of spatial attention in autism. J Neurosci. 2013;33:6776–81.
- 63.Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–88.
- 64.Rochira V, Carani C. Aromatase deficiency in men: a clinical perspective. Nat Rev Endocrinol. 2009;5:559–68.
- 65.Chen Z, Wang O, Nie M, Elison K, Zhou D, Li M, et al. Aromatase deficiency in a Chinese adult man caused by novel compound heterozygous CYP19A1 mutations: effects of estrogen replacement therapy on the bone, lipid, liver and glucose metabolism. Mol Cell Endocrinol. 2015;399:32–42.
- 66.Bejerot S, Eriksson JM, Bonde S, Carlstrom K, Humble MB, Eriksson E. The extreme male brain revisited: gender coherence in adults with autism spectrum disorder. Br J Psychiatry. 2012;201:116–23.
- 67.Lu S-F, Mo Q, Hu S, Garippa C, Simon NG. Dehydroepiandrosterone upregulates neural androgen receptor level and transcriptional activity. J Neurobiol. 2003;57:163–71.
- 68.Yeh S, Miyamoto H, Shima H, Chang C. From estrogen to androgen receptor: A new pathway for sex hormones in prostate. Proc Natl Acad Sci USA. 1998;95:5527.
- 69.Pohl A, Cassidy S, Auyeung B, Baron-Cohen S. Uncovering steroidopathy in women with autism: a latent class analysis. Mol Autism. 2014;5:27.
- 70.Auyeung B, Taylor K, Hackett G, Baron-Cohen S. Foetal testosterone and autistic traits in 18 to 24-month-old children. Mol Autism. 2010;1:11.
- 71.Lauritsen MB, Jørgensen M, Madsen KM, Lemcke S, Toft S, Grove J, et al. Validity of childhood autism in the Danish Psychiatric Central Register: findings from a cohort sample born 1990-1999. J Autism Dev Disord. 2010;40:139–48.
- 72.Underwood MA, Gilbert WM, Sherman MP. Amniotic fluid: not just fetal urine anymore. J Perinatol 2005;25:341–8.